What are some of the challenges with database builds for medical device studies, given the current regulatory requirements? What are the important items to be aware of? In this post, we will look at these and other questions, with a focus on data management and database build work to support medical device studies for approval in the United States and European Union.
Medical devices
Medical devices are an important part of healthcare. They run the spectrum from the common (e.g., bandages) to diagnostic tests, complex surgical interventions, and drug delivery mechanisms. Before a medical device can be available on the market, it needs to be shown to be safe and effective. This is accomplished through testing based on regulatory requirements for 510(k) applications (medium-risk device), PMA (high-risk device) applications, and investigational device exemption (IDE) approvals.
The FDA has the following 3 risk categories for medical devices: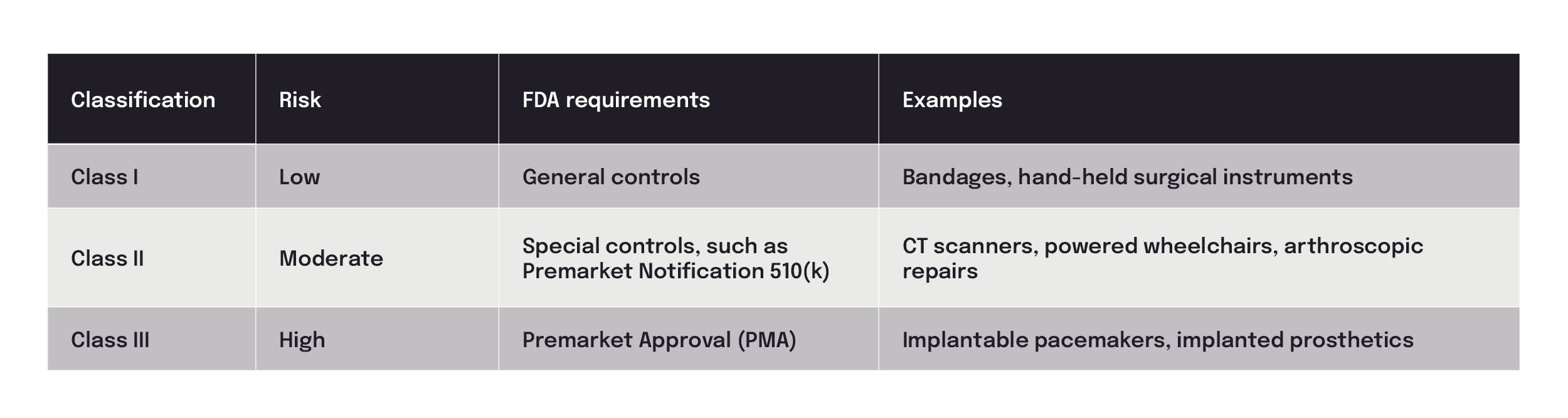
In addition to the FDA’s regulatory requirements, a recent requirement from the European Medicines Agency (EMA) of the EU, called the MDR Regulation (EU) 2017/745 on medical devices, went into effect in May 2021. This regulation was developed to improve the safety, reliability, and transparency of medical device information in the EU medical device market and has created a need for additional postmarketing studies and safety reporting requirements for medical devices.
Together, the FDA and EMA requirements present unique challenges for clinical trials and the necessary data for analyses to support the approval of medical devices.
Importance of database builds for medical device studies
A database build directly influences the quality of the study data and is an important part of a medical device study. The regulatory informational needs from both the FDA and EU need to be included in the overall strategy to build a quality, effective database.
Databases
The term “database” often refers to both the collected data as well as the database management system (DBMS) — or software that interacts with the end users, applications, and data.
Types of data sources include:
- Electronic case report form (eCRF) for electronic data collection (structured data), including device-specific identifiers (UDI-DD).
- Other electronic sources and documents, including unstructured data (e.g., X-ray, CT, MRI, device data, electronic participant-reported outcomes [ePRO], electronic clinical outcome assessments [eCOA]).
Database management systems may deal with:
- Web-based data entry.
- Query management.
- Data hosting (e.g., cloud platforms), data transfer, and data control access.
- Medical coding by importing coding dictionaries.
Database builds
The setup of a database includes planning for all data streams and may include strategies for skip logic, intelligent guided entry, and pop-ups to simplify the entry of data points following the ALCOA (Attributable, Legible, Contemporaneous, Original and Accurate) principles and to maintain GCP (Good Clinical Practice) principles for data reporting and data integrity. It must also be able to handle the substantial amounts of data found in many postmarket surveillance device studies.
Build challenges
The ability to integrate the various sources of data, from eCRF data to other electronic sources, into an effective clinical solution that is accessible and supports regulatory reporting requirements can be a challenge.
Choosing the right EDC (electronic data capture) system
Finding the right EDC system to meet the study needs and requirements at the right price point is needed for the satisfaction of both the sponsor and the data management team. Some important questions to choose the right EDC include:
- Will the trial have decentralized monitoring?
- Are advanced analytics and visualizations needed?
Working knowledge of the various EDC systems and technology to support needed capabilities is important.
Device-specific information challenges
Specific information on the device being investigated is needed for reporting purposes.
Although device trials tend to have smaller sample sizes than drug trials, the growing need for postmarketing studies, which typically involve long-term data collection, can dramatically increase the need for a database to support a large volume of data.
In 2013, the FDA published the final rule for the Unique Device Identification (UDI) System requiring manufacturers to label marketing devices with a UDI. The UDI-DD identifies the manufacturer and model as well as the lot number, serial number, and expiration date. This increases the traceability of the device and implant identification and supports reporting for adverse events (AEs), recalls, and other surveillance measures for reporting.
Some FAQs for medical device database builds by Veranex
What kinds of device trials have you supported?
As a CRO specializing in medical devices, we have supported database builds for Class I, II, and III trials. Class I trials have the fewest requirements of all trials, requiring only a study protocol and source document for the few PRO forms typically used. A Class III trial (PMA) requires the most extensive effort, needing systems designed to capture secondary and tertiary endpoint information.

What kind of targeted assistance can Veranex provide for a device study database build?
Focusing on the uniqueness of device trials, as well as common pitfalls, efforts could include:
- Expertise on the needed data points based on the regulatory requirements of FDA, EMA, or another agency.
- Planning of a database design that can also support the multiple protocol amendments often found in long-term studies. Because of enrollment challenges or other changes to the study, additional time or visits may be needed as a post-production change to the database. Instead of creating static visits, the initial database can be designed with flexibility to add visits when required. In this way, additional efforts, along with costly change orders, are minimized.
- Customizing the visibility/entry of the eCRF based on study role (e.g., independent assessor, which is often present in device trials). For example, to facilitate site-specific device safety assessments by the principal investigator (PI), data entry for ONLY the participants for the respective site can be made accessible via the PI Assessment form, and the same form is not enabled for participants at other sites.
- Customizing the design for bilateral cases to support the site’s ability to enroll participants in bilateral cases (i.e., if the participant has an indication in both legs), then both legs will be involved in the study with the ability to collect different sets of data with the same participant number.
- Providing an end user–friendly database that allows the site to select and enter data easily instead of having to complete more tedious manual entry.
- Efficient entry of device numbers (catalog numbers/lot numbers/device identifiers) by loading a catalog or schema with device numbers or lot numbers already generated. Through APIs (application programming interface), this information can be fed into an IRT system to be integrated into the EDC system. Every time a device is assigned to a participant in the trial, a site can select the number in the IRT system, and that number is integrated into the database.
- Lab set-up for easier lab data collection, by creating eCRF forms with a list of analytes, units, and reference ranges for each lab across all sites. Local lab results can be entered into the system for each lab analyte, then preconfigured conversion factors normalize the data into a common standard unit, in real time. Normal reference range values for each lab test, with gender and age attributes, can be maintained, and the system can send an alert for out-of-range values.
- Safety traceability by customizing the sending of safety alerts (serious AE [SAE] and device deficiency) to the respective stakeholders with the necessary information so quick action can be taken by the safety team.
- Different ePRO regulatory versions to support multiple sites and countries. Libraries can be maintained to handle these versions and dynamically enable the ePRO at the respective sites to avoid confusion.
In what areas can cost efficiencies be found?
- In most EDC systems, safety data trackers can be built for AE and device deficiency reporting, eliminating the need for additional efforts.
- Email notification for multiple scenarios that can be used for the immediate reporting of AEs/device deficiencies to regulatory bodies can be programmed within an EDC system.
- Efficiency can also be gained by using previous study designs and data trends, which will yield faster and less expensive studies.
- A franchise approach supports tailoring of the CRF and database builds for a single sponsor to meet the needs of multiple products and involves the following:
- Use of the global library to create different CRF versions for the same forms across different product teams
- Drawing from a library of ePROs, which can be generated based on regional differences
- Design of a single database to cover multiple sites with different regulatory standards
These challenges are different from those encountered to implement the processes and data needs of a clinical trial for a drug and are best met with the expertise of professionals who have built databases for device trials based on relevant regional experience.
Veranex delivers end-to-end integrated services with a wealth of expertise in medical device solutions, including data management and analytics services. We are here to help. For more information, contact us.