



 Neeru Mittal, PhD
Neeru Mittal, PhD
The landscape of modern healthcare has been completely transformed by the development of cutting-edge medical devices. From life-saving implants to innovative surgical tools, these medical devices have become indispensable in healthcare. However, behind every successful medical device lies a crucial consideration that forms one of the cornerstones of its success: biocompatibility.
What is the Biocompatibility of Medical Devices?
ISO 10993-1:2018, the guiding standard, defines biocompatibility as the ability of a medical device or material to perform with an appropriate host response in a specific application. In essence, it denotes the device’s ability to seamlessly interact with the human body without causing harm or triggering adverse reactions.
In this blog post, we explore the intricacies of biocompatibility for medical devices, shedding light on the biological risk assessment process following ISO 10993-1. We also look into the hot topics and trends that urgently need your attention or can be an important tool as you assess the biological safety of your medical device.
ISO 10993 Series
ISO 10993 series is a set of international standards that provides a comprehensive framework for evaluating the biological safety of medical devices. Three key standards within this series, ISO 10993-1, ISO 10993-17, and ISO 10993-18, stand as pivotal contributors to the meticulous process of assessing potential risks associated with medical devices.
Understanding Biological Risk Assessment per ISO 10993-1
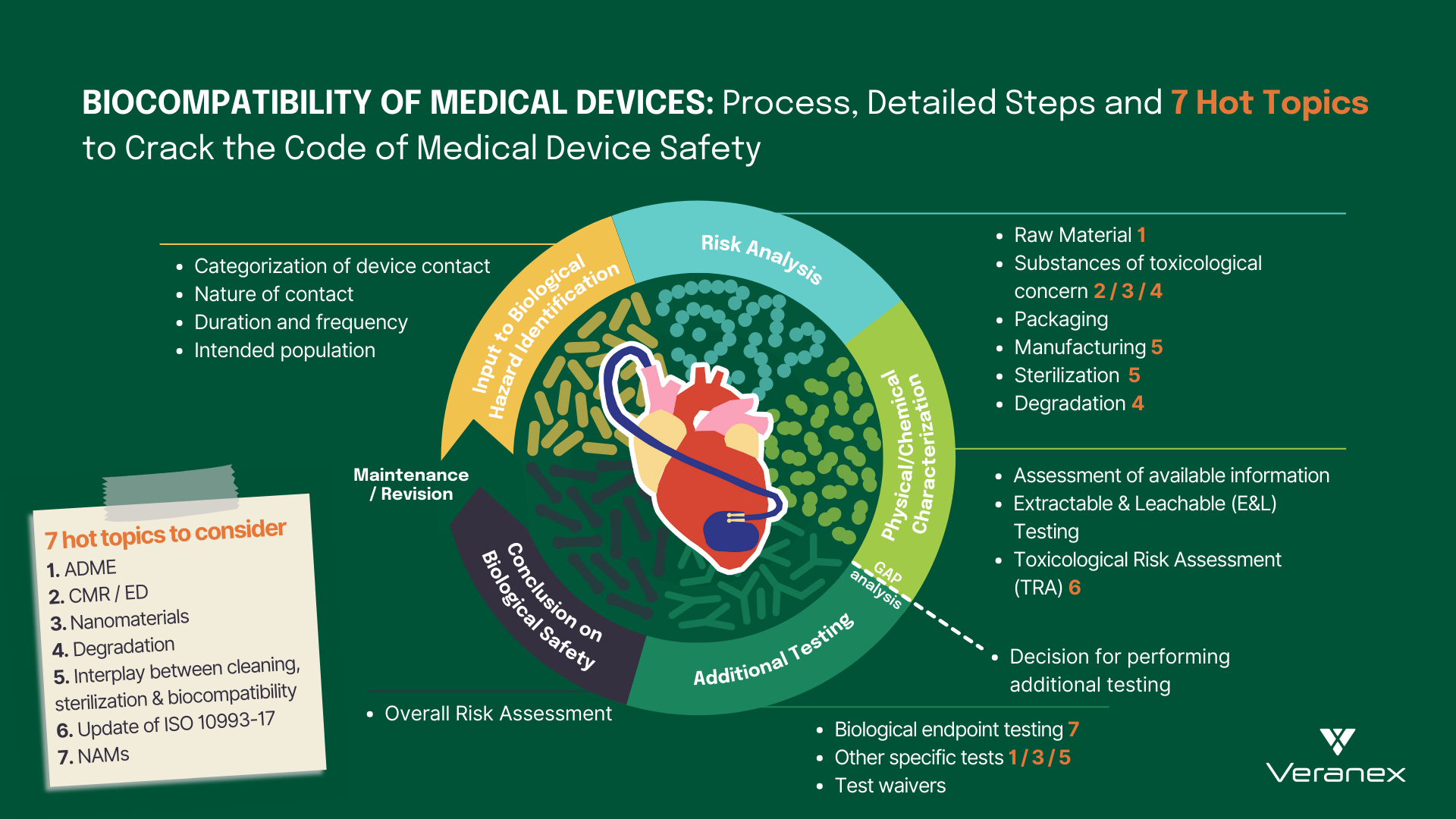
Biological risk assessment of medical devices, as outlined in ISO 10993-1:2018, involves a systematic and comprehensive analysis of potential biological hazards associated with the use of the medical device.
The journey of biological risk assessment begins with the categorization of the medical device according to its intended use and contact duration with the human body. Is your medical device surface-contacting, externally communicating, or an implant? Does it establish direct or indirect contact with the patient? This device categorization leads us to Table A.1 in ISO 10993-1:2018 which provides a list of biological endpoint tests that must be considered during the biological risk assessment.
Following this, a theoretical chemical characterization unfolds, wherein a comprehensive evaluation of the device’s raw materials, packaging materials, manufacturing processes, additives, and potential sterilization techniques is conducted to identify possible chemical constituents that could elicit adverse biological responses. This information is gathered from a variety of safety databases and different documents such as material formulation disclosures, certificates of analysis, compliance statements, technical datasheets, and safety data sheets. The evaluation process does not stop here, as the risk assessment continues further into understanding device degradation during storage, clinical use, and if applicable, reprocessing phases. This approach encompasses the entire life cycle of the device.
The collected data is then construed within the context of existing medical knowledge to estimate the biological risks posed by the device. In light of these risks, an assessment of the chemical and biological testing that is critical for the device, with information sourced from Table A.1 in ISO 10993-1:2018, is undertaken. Notably, performing chemical characterization testing, sometimes including chemical testing also referred to as extractable and leachable (E&L) testing, has become the norm in the present days. Chemical characterization together with emerging New Approach Methodologies (NAMs) testing is embraced for its ability to circumvent unnecessary animal testing, thus respecting the animal welfare principles of ISO 10993-2.
ISO 10993-18:2020 provides insights into the execution of leachable/extractable studies, which then unveils what compounds could possibly migrate from a medical device into the body. Techniques such as gas or liquid chromatography (GC or LC), mass spectrometry (MS), and inductively coupled plasma spectroscopy (or coupled with MS) are employed to unveil the intricate molecular composition of the medical device extract. Post this analysis, a toxicological risk assessment following ISO 10993-17 is performed to confirm the absence of substances leaching from the device in quantities of toxicological concern. The findings of this assessment pave the way for the subsequent steps detailed in ISO 10993-1. In essence, the outcomes of the chemical testing normally dictate the need for specific biological tests to mitigate all the possible risks for the assessed device.
This pragmatic approach/methodology ensures a robust and standardized approach to evaluating the biological safety of medical devices, safeguarding patient well-being and facilitating regulatory compliance.

Hot Topics and Trends
With the transition to the Medical Device Regulation (MDR) in Europe, manufacturers have undertaken a thorough revision of their biological risk assessments. Over the last few years, Veranex has collected feedback from various Notified Bodies and identified specific topics that often are under focus. Through our participation as experts in ISO/TC194, we have also gathered important insights into the updates of the ISO 10993 standards. These hot topics and updates, warranting your immediate attention, include but are not restricted to the following:
1. ADME: Manufacturers of substance-based medical devices, medical devices that achieve their intended purpose by the action of their ingredients, are faced with several challenges as the MDR presents new requirements for these devices. Some of the requirements are the documentation of pharmacokinetics; absorption, distribution, metabolism and excretion (ADME) that is needed to address safety concerns and also to correctly classify the medical devices. Many of these devices can also be borderline products and hence need to show scientific evidence that their principal mode of action is physical, chemical or mechanical to correctly classify the product as a medical device.
2. CMR/ED: There is a growing focus directed towards appraising Carcinogenic, Mutagenic, and Reprotoxic (CMR) substances as well as endocrine-disrupting (ED) substances, within medical devices. According to MDR guidelines, the presence of CMR/ED substances exceeding 0.1 wt.% requires thorough quantification and justification, highlighting the need for holistic CMR/ED risk assessments.
3. Nanomaterials: There is a heightened emphasis currently being put on the assessment of nanomaterials within the biological assessment of medical devices. This pertains to both devices containing nanomaterials and those with the potential to generate nanomaterials through processes like degradation, wear, or mechanical interactions. This increased focus underscores the need for a structured approach to comprehending nanomaterials’ effects within the scope of biological assessments.
4. Degradation: A medical device is susceptible to degradation at any stage of its life cycle. Therefore, a thorough evaluation determining the need for degradation testing, appropriate testing methods, and meticulous analysis of results becomes imperative. Relevant standards within the ISO 10993 series including ISO 10993-9, ISO 10993-13, ISO 10993-14, and ISO 10993-15 must be considered for this analysis.
5. Interplay between cleaning validation, sterilization validation, and biocompatibility: Testing optimizations are sometimes possible when sterilization, cleaning and biocompatibility are considered simultaneously and early enough. Current standard developments are being made in order to plan, establish, and optimize Process Validation and Verification and Validation activities.
6. ISO 10993-17 Update: The recent release of ISO 10993-17:2023 is set to bring forth many novel concepts such as establishing a framework for key notions like screening data using Toxicological Screening Limit (TSL), deriving Tolerable Intake (TI) and Tolerable Contact Levels (TCL), and calculating Margin of Safety (MOS). It also advocates the use of release kinetics data to support TRA analysis of medical devices, particularly for long-term contacting devices. Manufacturers are advised to anticipate and integrate these changes into their device’s biological risk assessment process.
7. NAMs: Medical device toxicology is currently undergoing an exciting evolution with an increased focus on the use of New Approach Methodologies (NAMs) including in vitro methods for the safety assessment of medical devices. The use of NAMs for specific endpoints has already gained regulatory acceptance in large parts of the world and provides a more human-relevant prediction for the risk assessment of medical devices.
We have the expertise needed for your biological risk assessment.
Veranex has a dedicated, highly experienced team that combines both regulatory and technical expertise and is available to support you in setting up or reviewing your Biological Risk Assessment. Working with us, you can be sure that your documentation follows the best practices concerning the hot topics mentioned in this article, and that any questions from regulatory authorities are answered in the fastest and most efficient way.
By Dr. Neeru Mittal and Dr. Rose-Marie Jenvert, biocompatibility experts at Veranex.
